
HET ONDERWERP IN HET KORT
• Sommige aspecten van de microbiologie blijven mysterieus vanwege een gebrek aan informatie over de identiteit en rol van veel microbiële genen en eiwitten.
• Het vermogen om microbiële sequenties op schaal en tussen soorten te verkrijgen en te analyseren, inclusief die welke niet onder laboratoriumomstandigheden kunnen worden gekweekt, levert inzichten en gegevens op om te onderzoeken.
• Inschrijven NatuurRodríguez del Río et al.1 rapporteren hun analyse van 149.842 bacteriële genomen die zijn bemonsterd uit verschillende habitats in het wild.
• De gegevens werden gebruikt om sequenties te selecteren om een catalogus van 404.085 voorheen onbekende genfamilies te genereren die prioriteit konden krijgen voor verder onderzoek.
• Het onderzoek naar deze voorheen onbekende genen zou kunnen leiden tot nieuwe klinische instrumenten of nieuwe perspectieven kunnen bieden over hoe micro-organismen evolueerden om te overleven in hun natuurlijke omgeving.
JAKOB WIRBEL & AMI S. BHATT: Structuur en context brengen in genmysteries
De functie van de meeste microbiële genen is onbekend. Een deel van deze microbiële ‘donkere materie’ codeert mogelijk voor voorheen onbekende typen enzymen of antibioticaklassen. Naarmate er steeds meer genen met een onbekende functie worden ontdekt door het sequencen van DNA uit mengsels van meerdere genomen, ook wel metagenomische sequencing genoemd, heeft de moeilijkheid van het experimenteel karakteriseren van deze enigmatische genen geleid tot een focus op het computationeel voorspellen van hun functie.2. Twee publicaties in Natuuréén van Rodríguez del Río et al.1en één van Pavlopoulos et al.3 die afgelopen oktober werd gepubliceerd, deze uitdaging aanpakken door slim gebruik te maken van de vooruitgang in clusteralgoritmen (computationele hulpmiddelen die genen groeperen op foundation van overeenkomsten in de aminozuursequentie) en hulpmiddelen voor het voorspellen van de eiwitstructuur4 zoals AlphaFold.
Lees het artikel: Functionele en evolutionaire betekenis van onbekende genen uit niet-gecultiveerde taxa
Ondanks verschillende technische benaderingen is de kernstrategie die door Pavlopoulos wordt gebruikt et al. en Rodríguez del Río et al. was vergelijkbaar. Beiden clusterden honderden miljoenen eiwitsequenties uit metagenomische datasets in voorheen onbekende eiwitfamilies. Rodríguez del Río en collega’s filterden hun gegevens om alleen genen van prokaryoten (organismen waarvan de cellen een kern missen) te onderzoeken, terwijl Pavlopoulos et al. gebruikte gegevens die ook sequenties van eukaryoten (organismen waarvan de cellen een kern hebben) en virussen bevatten.
Met deze catalogi van voorheen onbekende households bij de hand, wilden beide groups de functie van hun nieuw beschreven households voorspellen, gebruikmakend van genomische contextanalyse, waarbij aangrenzende genen worden onderzocht op aanwijzingen over de functie, en doorbraken worden benut in methoden om te voorspellen eiwitstructuren. In prokaryote genomen zijn genen die bij dezelfde route betrokken zijn vaak dicht bij elkaar aanwezig. Genomische contextanalyse, die ‘schuld door associatie’ voorstelt, is effectief gebruikt om voorheen onbekende antivirale afweersystemen te voorspellen die door bacteriën worden gebruikt5. De tweede benadering, het vergelijken van voorspelde eiwitstructuren om vergelijkbare (homologe) eiwitten te vinden, is gevoeliger dan alleen het vergelijken van aminozuursequenties alleen.6. Beide groups voorspelden structuren voor hun eiwitfamilies en vergeleken deze met databases van bekende structuren, waardoor weloverwogen voorspellingen werden gedaan over de functie van enkele van deze raadselachtige eiwitten.
De enorme omvang en de computerinvesteringen die bij deze inspanningen betrokken zijn, die honderdduizenden nieuw ontdekte eiwitfamilies opleverden (Fig. 1), zijn indrukwekkend. Toch blijft het aantal voorheen onbekende genen met een functionele voorspelling nog steeds relatief klein. In beide publicaties kon slechts ongeveer 15% van de voorheen onbekende eiwitfamilies worden geannoteerd op foundation van structurele gelijkenis; genomische contextanalyse maakte het mogelijk om functies voor te stellen voor 7,4% van de households in Pavlopoulos et al. en 13% in Rodríguez del Río en collega’s. Bovendien missen sommige toegewezen functionele categorieën (zoals ‘ribosoom’) een gedetailleerde specificiteit en dit zou de precieze rol van deze genen kunnen verdoezelen. Uiteindelijk zal de betrouwbaarheid van deze voorspellingen experimenteel moeten worden vastgesteld. Rodríguez del Río inderdaad et al. zette de eerste stap op weg naar dit doel door de annotatie voor twee van hun voorspelde households experimenteel te verifiëren.

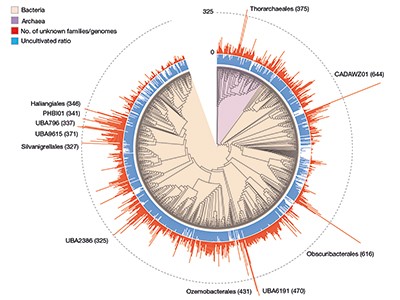
Figuur 1 | Voorheen onbekende microbiële genfamilies. De grootschalige analyse van DNA-sequenties gevangen uit microbiële monsters, zoals gerapporteerd door Rodríguez del Río et al.1 en door Pavlopoulos et al.3 heeft honderdduizenden voorheen onbekende genfamilies onthuld. Deze gegevens – die zijn verzameld van microben in het wild en in verschillende habitats, en die soorten omvatten die niet in het laboratorium zijn gekweekt – bieden een startpunt voor het verkrijgen van inzicht in onontdekte aspecten van de biologie van bacteriële en archaeale micro-organismen. Figuur aangepast van figuur 3a van ref. 1.
Door dieper in de microbiële donkere materie te duiken, ontsluiten deze twee onderzoeken een schat aan voorheen verborgen kennis, waardoor de weg wordt vrijgemaakt voor toekomstige ontdekkingen op various gebieden, van geneeskunde tot biotechnologie. Vervolgexperimenten zouden de studie van eiwitfamilies met volledig nieuwe eiwitvouwen kunnen omvatten, waardoor mogelijk onontgonnen biologische functies aan het licht komen. Op dezelfde manier kunnen synapomorfe genen – die overeenkomen met eiwitfamilies die specifiek zijn voor een groep organismen die een gemeenschappelijke voorouder delen, maar afwezig zijn in andere – aanwijzingen bevatten voor belangrijke evolutionaire processen. Met verdere verfijning en validatie bieden deze computationele benaderingen een krachtig hulpmiddel voor het ontsluiten van de functionele geheimen van de onzichtbare microbiële wereld.
ALEXANDER J. PROBST: Microbiële sequenties onthullen ecologie en evolutie
Genen zijn de ultieme bron van alle biologische informatie op aarde, van de kleur van het menselijk oog tot de celvorm van micro-organismen. De eiwitten waarvoor ze coderen kunnen met behulp van bio-informatica in households worden gegroepeerd, meestal met gedeelde functionaliteit. Het geheel van alle bekende eiwitten in databases breidt zich voortdurend uit naarmate de sequentie van genomen wordt bepaald en de functies van de gecodeerde eiwitten worden voorspeld. Het grootste deel van de biologische functionele diversiteit op onze planeet wordt toegeschreven aan microbiële eiwitten. Met de komst van de sequencing van gemengde microbiële genomen uit de omgeving (een aanpak die meerdere genomen onderzoekt en metagenomics wordt genoemd7), is de toename in de snelheid waarmee gegevens worden toegevoegd aan genoom- en eiwitdatabases opvallend. De functionele capaciteit van de meeste eiwitfamilies is echter onbekend en maakt deel uit van de microbiële donkere materie.
Het volgen van mensen en microben
Het werk van Rodríguez del Río en collega’s, evenals de studie van Pavlopoulos et al., analyseerde grootschalige metagenomische gegevens en onderzocht de potentiële functie en distributie van onbekende eiwitfamilies, die evolutionair en ecologisch belang zouden kunnen hebben. Rodríguez del Río analyseerde bijna 150.000 microbiële genomen (Fig. 1), en Pavlopoulos en collega’s onderzochten bijna 27.000 metagenomische datasets afkomstig uit various ecosystemen met verschillende bio-informatica-benaderingen – die veel verder gaan dan de schaal van gegevens in openbare databases die in eerdere dergelijke onderzoeken zijn gebruikt.8. Verrassend genoeg onthulde een methode genaamd verdunningsanalyse, gebruikt door Pavlopoulos en collega’s, geen vertraging in de detectie van voorheen onbekende eiwitfamilies toen nieuwe metagenomen aan hun analyse werden toegevoegd. In plaats daarvan nam de detectie van eiwitfamilies exponentieel toe, wat een reeks vervolgstudies rechtvaardigde.
De verdeling van eiwitfamilies over de ecosysteemcategorieën (biomen) van de aarde, gepresenteerd door Pavlopoulos en collega’s, bevestigt de bevindingen van eerdere onderzoeken met betrekking tot de verspreiding van microbiële genen.8. Sommige biologische entiteiten waren echter bijzonder rijke bronnen van nieuw ontdekte eiwitfamilies, waaronder virussen, zoals Pavlopoulos et al. rapport, en microben genaamd Asgardarchaeota, zoals gepresenteerd door Rodríguez del Río en collega’s. Deze laatste zijn een groep micro-organismen genaamd archaea die nauw verwant zijn aan de eerste voorouder van eukaryoten. Als zodanig zou het bestuderen van hun eiwitten nieuwe inzichten kunnen onthullen in de evolutie van de eukaryotische cel9.
Crowdsourcing van de microben op aarde
Een grote uitdaging bij het onderzoeken van de rijkdom aan voorheen onbekende eiwitfamilies gecodeerd in genomen van natuurlijke monsters is de identificatie van eukaryote genen in metagenomen. Hoewel er bepaalde algoritmen bestaan voor het terugwinnen van eukaryote genomen uit metagenomen, is het nauwkeurig voorspellen van eukaryote genen in gemengde DNA-sequenties – gelijkwaardig aan de methode van Pavlopoulos en collega’s om microbiële genen te identificeren – bio-informatisch nog steeds niet mogelijk. Zodra deze tekortkoming is overwonnen met de ontwikkeling van nieuwe algoritmen, zullen wetenschappers de ‘sequentieruimte’ van eiwitten substantieel uitbreiden en eiwitfamilies met een onbekende functie identificeren die de ecologie en evolutie van eukaryoten aansturen.
De grootste vooruitgang bij het nauwgezet organiseren van de eiwitfamilies van bijna 27.000 metagenomen en in de levensboom ligt in de identificatie van ecosysteemspecifieke eiwitclusters die verschillen in termen van hun aan- of afwezigheid, of relatieve overvloed tussen verschillende omstandigheden van een bepaald ecosysteem – bijvoorbeeld tussen de contexten van gezondheid of ziekte. Door deze strategie toe te passen om microbiële gegevens voor gezonde mensen en mensen met colorectale kanker te onderzoeken, ontdekten Rodríguez del Río en collega’s dat specifieke onbekende eiwitfamilies verrijkt waren in de darmbacteriën van mensen met kanker. Deze eiwitfamilies werden geassocieerd met microbiële motiliteit, adhesie en potentiële invasie van menselijk weefsel, zoals onthuld door genomische contextanalyse. Het benutten van deze aanpak in andere onderzoeksgebieden zou uiterst nuttig moeten zijn voor het ontcijferen van de verschillende functies van monstersets, in de hoop nieuwe doelen voor biochemische analyses te identificeren om licht te werpen op een klein deel van de microbiële donkere materie.
Het identificeren van verschillen in microbiële gemeenschappen (microbiomen) die bijvoorbeeld de ziektetoestand van een persoon kunnen verklaren, sterk afhankelijk zijn van het vergelijken van welke soorten aanwezig zijn en hoe overvloedig ze zijn (de taxonomische samenstelling), en het onderzoeken van genen die geassocieerd zijn met bepaalde functies . Het vinden van specifieke maar differentieel overvloedige eiwitfamilies met een onbekende functie, zoals aangetoond door Rodríguez del Río en collega’s, heeft het potentieel om niet alleen de huidige, op markergenen gebaseerde benaderingen voor het differentiëren van microbiomen te vervangen, maar ook om het microbioomonderzoek naar een nieuw en causaal niveau te brengen. -gedreven niveau.